Cell-Killing Proteins Suppress Listeria Without Killing Cells
For Immediate Release
New North Carolina State University research shows that key proteins known for their ability to prevent viral infections by inducing cell death can also block certain bacterial infections without triggering the death of the host cells.
Rather than killing host cells infected by Listeria in the gastrointestinal tract, the RIPK3 and MLKL proteins recognize the chemical composition of the bacteria and MLKL binds to it, preventing the spread of Listeria while keeping the host cells alive.
“While we’ve shown that these proteins take on a different function in intestinal epithelial cells than they do in immune cells, we’re still not sure how or why this differentiation occurs,” said Jun Ninomiya-Tsuji, professor of biological sciences and co-corresponding author of a paper describing the research.
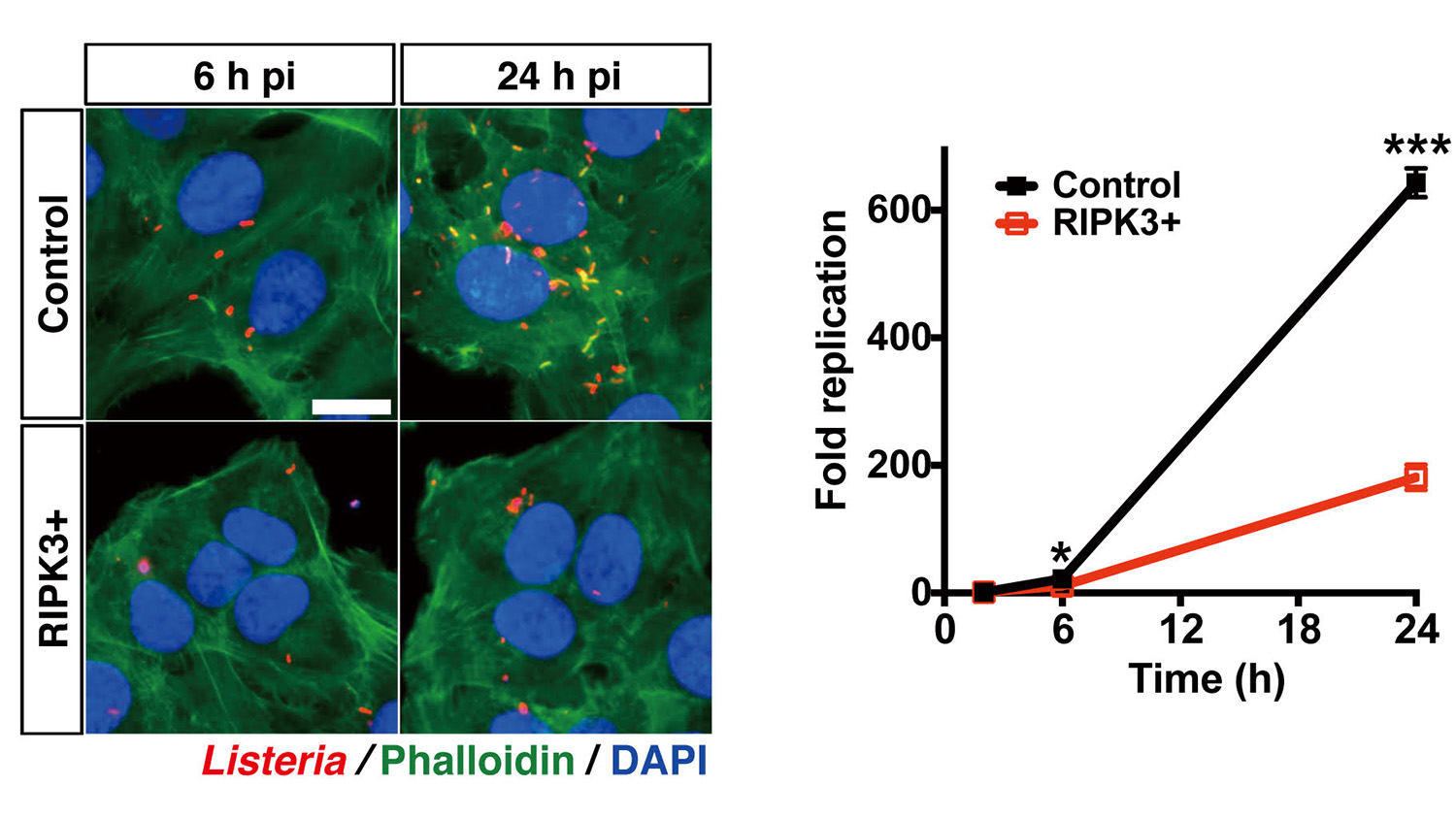
The researchers, led by Kazuhito Sai, a toxicology research associate and co-corresponding author of the paper, first used human intestinal cells to show that RIPK3-deficient cells were infected by Listeria while cells with RIPK3 had few such infections. The researchers then used mice to see if Listeria could reach mouse livers by invading intestinal cells. They found many Listeria in RIPK3-deficient mice but few Listeria in normal mice.
They then showed that RIPK3 and a protein that works with it, MLKL, were activated by the presence of Listeria. This protein-pathway activation inhibited Listeria replication, showing that the proteins effectively blunted Listeria.
Next, and most surprisingly, the researchers showed that the activation of RIPK3 and MLKL by Listeria did not result in cell death. Instead, MLKL proteins bound themselves to Listeria, stopping its spread.
“These proteins induce cell death to prevent certain infections, particularly in immune cells,” Sai said. “Inducing death of epithelial cells in the GI tract may cause removal of an important barrier to viruses and bacteria, so it’s possible that these proteins recognize that killing these cells could make things worse instead of better.”
Future research will attempt to understand how and why these proteins take different approaches – inducing cell death or not – to stave off bacteria in the GI tract, the researchers said.
Postdoctoral researcher Cameron Parsons, bioinformatics research scholar John S. House and Sophia Kathariou, professor of food, bioprocessing and nutrition sciences, co-authored the paper, which was published online in the Journal of Cell Biology. The National Institutes of Health funded the study under grants GM112986 and P30ES025128. Confocal microscopy imaging was partly supported under National Science Foundation grant DBI-1624613.
-kulikowski-
Note to editors: An abstract of the paper follows.
“Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing”
Authors: Kazuhito Sai, Cameron Parsons, John S. House, Sophia Kathariou, Jun Ninomiya-Tsuji, North Carolina State University
Published: April 11, 2019 in the Journal of Cell Biology
DOI: 10.1083/jcb.201810014
Abstract: RIPK3, a key mediator of necroptosis, has been implicated in the host defense against viral infection primary in immune cells. However, gene expression analysis revealed that RIPK3 is abundantly expressed not only in immune organs but also in the gastrointestinal tract, particularly in the small intestine. We found that orally inoculated Listeria monocytogenes, a bacterial foodborne pathogen, efficiently spread and caused systemic infection in Ripk3-deficient mice while almost no dissemination was observed in wild-type mice. Listeria infection activated the RIPK3-MLKL pathway in cultured cells, which resulted in suppression of intracellular replication of Listeria. Surprisingly, Listeria infection–induced phosphorylation of MLKL did not result in host cell killing. We found that MLKL directly binds to Listeria and inhibits their replication in the cytosol. Our findings have revealed a novel functional role of the RIPK3-MLKL pathway in nonimmune cell-derived host defense against Listeria invasion, which is mediated through cell death–independent mechanisms.
- Categories:


