Researchers Use Molecular Dynamics and Machine Learning to Create ‘Hyper-predictive’ Computer Models for Drug Discovery
For Immediate Release
Researchers from North Carolina State University have demonstrated that molecular dynamics simulations and machine learning techniques could be integrated to create more accurate computer prediction models. These “hyper-predictive” models could be used to quickly predict which new chemical compounds could be promising drug candidates.
Drug development is a costly and time-consuming process. To narrow down the number of chemical compounds that could be potential drug candidates, scientists utilize computer models that can predict how a particular chemical compound might interact with a biological target of interest – for example, a key protein that might be involved with a disease process. Traditionally, this is done via quantitative structure-activity relationship (QSAR) modeling and molecular docking, which rely on 2- and 3-D information about those chemicals.
Denis Fourches, assistant professor of computational chemistry, wanted to improve upon the accuracy of these QSAR models. “When you’re screening a set of 30 million compounds, you don’t necessarily need a very high reliability with your model – you’re just getting a ballpark idea about the top 5 or 10 percent of that virtual library. But if you’re attempting to narrow a field of 200 analogues down to 10, which is more commonly the case in drug development, your modeling technique must be extremely accurate. Current techniques are definitely not reliable enough.”
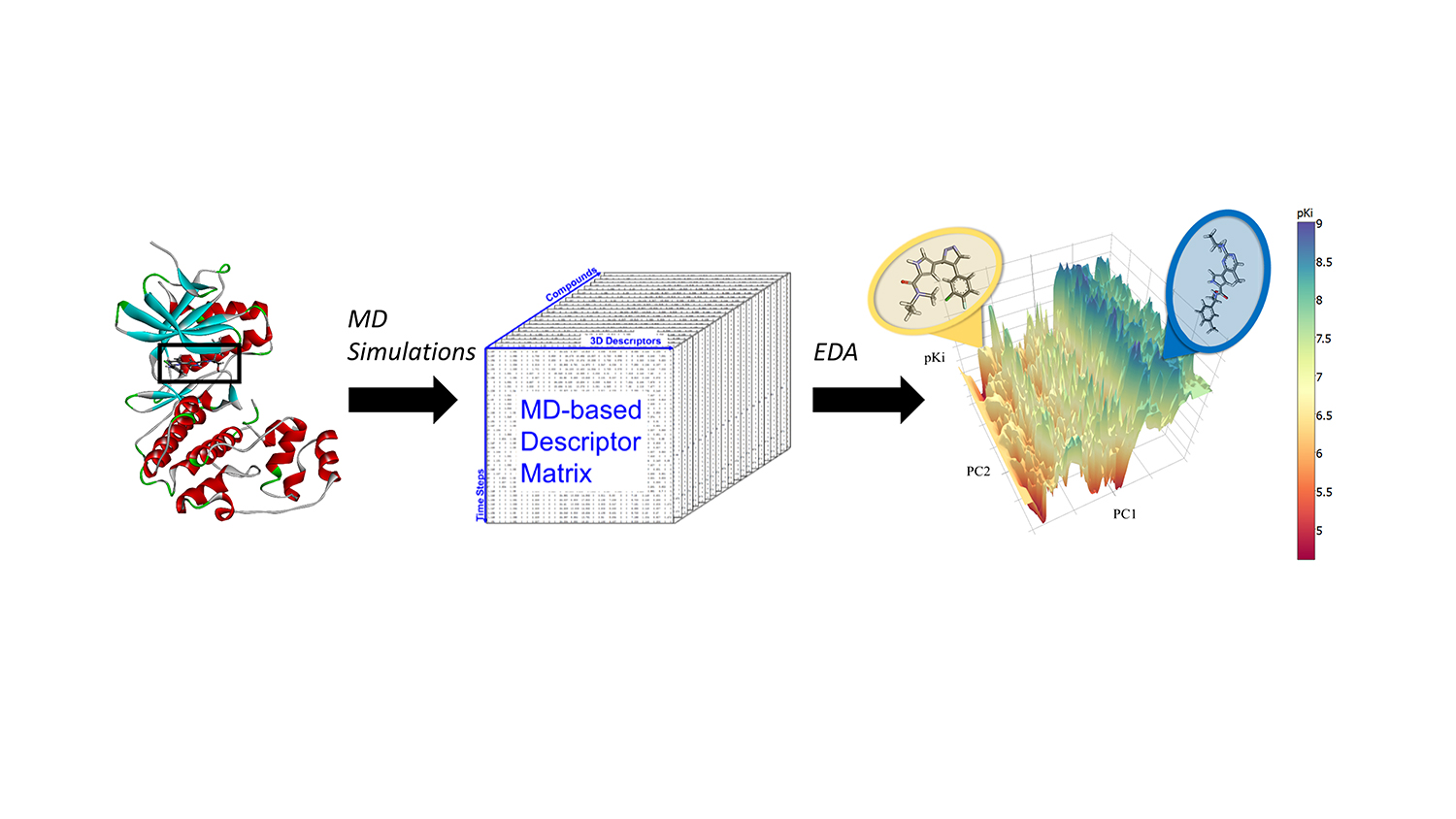
Fourches and Jeremy Ash, a graduate student in bioinformatics, decided to incorporate the results of molecular dynamics calculations – all-atom simulations of how a particular compound moves in the binding pocket of a protein – into prediction models based on machine learning.
“Most models only use the two-dimensional structures of molecules,” Fourches says. “But in reality, chemicals are complex three-dimensional objects that move, vibrate and have dynamic intermolecular interactions with the protein once docked in its binding site. You cannot see that if you just look at the 2-D or 3-D structure of a given molecule.”
In a proof-of-concept study, Fourches and Ash looked at the ERK2 kinase – an enzyme associated with several types of cancer – and a group of 87 known ERK2 inhibitors, ranging from very active to inactive. They ran independent molecular dynamics (MD) simulations for each of those 87 compounds and computed critical information about the flexibility of each compound once in the ERK2 pocket. Then they analyzed the MD descriptors using cheminformatics techniques and machine learning. The MD descriptors were able to accurately distinguish active ERK2 inhibitors from weakly actives and inactives, which was not the case when the models used only 2-D and 3-D structural information.
“We already had data about these 87 molecules and their activity at ERK2,” Fourches says. “So we tested to see if our model was able to reliably find the most active compounds. Indeed, it accurately distinguished between strong and weak ERK2 inhibitors, and because MD descriptors encoded the interactions those compounds create in the pocket of ERK2, it also gave us more insight into why the strong inhibitors worked well.
“Before computing advances allowed us to simulate this kind of data, it would have taken us six months to simulate one single molecule in the pocket of ERK2. Thanks to GPU acceleration, now it only takes three hours. That is a game changer. I’m hopeful that incorporating data extracted from molecular dynamics into QSAR models will enable a new generation of hyper-predictive models that will help bringing novel, effective drugs onto the market even faster. It’s artificial intelligence working for us to discover the drugs of tomorrow.”
The work appears in the Journal of Chemical Information and Modeling. Ash is first author of the paper and is funded by an NIEHS grant (T32ES007329). Other funding was provided by the NC State Chancellor’s Faculty Excellence Program.
-peake-
Note to editors: An abstract of the paper follows.
“Characterizing the Chemical Space of ERK2 Kinase Inhibitors Using Descriptors Computed from Molecular Dynamics Trajectories”
DOI: 10.1021/acs.jcim.7b00048
Authors: Jeremy Ash, Denis Fourches, North Carolina State University
Published: Journal of Chemical Information and Modeling
Abstract:
Quantitative Structure-Activity Relationship (QSAR) models typically rely on 2D and 3D molecular descriptors to characterize chemicals and forecast their experimental activities. Previously, we showed that even the most reliable 2D QSAR models and structure-based 3D molecular docking techniques were not capable of accurately ranking a set of known inhibitors for the ERK2 kinase, a key player in various types of cancer. Herein, we calculated and analyzed a series of chemical descriptors computed from the molecular dynamics (MD) trajectories of ERK2-ligand complexes. First, the docking of 87 ERK2 ligands with known binding affinities was accomplished using Schrodinger’s Glide software; then, solvent-explicit MD simulations (20 ns, NPT, 300K, TIP3P, 1fs) were performed using the GPU-accelerated Desmond program. Second, we calculated a series of MD descriptors based on the distributions of 3D descriptors computed for representative samples of the ligand’s conformations over the MD simulations. Third, we analyzed the dataset of 87 inhibitors in the MD chemical descriptor space. We showed that MD descriptors (i) had little correlation with conventionally used 2D/3D descriptors, (ii) were able to distinguish the most active ERK2 inhibitors from the moderate/weak actives and inactives, and (iii) provided key and complementary information about the unique characteristics of active ligands. This study represents the largest attempt to utilize MD-extracted chemical descriptors to characterize and model a series of bioactive molecules. MD descriptors could enable the next generation of hyper-predictive MD-QSAR models for computer-aided lead optimization and analogue prioritization.
- Categories:


